Power Up. Energy (ATP) is produced in mitochondria by enzymes called ATP synthetases embedded in the inner membrane. In this animation, travel to the inner membrane and see ATP (glowing orange) being made and spent, forming ADP (dull yellow). Be sure to take a look at all the proteins on the way. Video: XVIVO for Harvard University.
Mitochondria produce the energy needed to keep muscles healthy and moving. But in people with ALS, these power plants go out of service likely contributing to muscle atrophy and ultimately, paralysis.
Scientists are developing treatments to supe up mitochondria in hopes to keep the energy flowing in the muscles and connecting nerves. One of these emerging medicines, Knopp Biosciences’ dexpramipexole now licensed by Biogen Idec, is gathering steam as a potential ALS treatment strategy. Reporting phase II results last fall, neurologists found that the drug slowed disease progression about 30%.
Some researchers suspect however, that these mitochondrial-targeted medicines may need to do much more than boost energy production to grind ALS to a halt. Even operating at full steam, these power plants may be unable to provide enough energy to keep muscles working because they may not be in the right place to do their job. Scientists are now beginning to understand why these mitochondria might stray, suggesting new strategies to tackle the disease.
Traffic Tie-up
When Massachusetts General Hospital researchers reported the first altered gene, superoxide dismutase 1 (SOD1), linked to ALS in 1993, researchers scrambled to generate and characterize mice with these same mutations in hopes to discover the cause of the disease.
Reporting the first mouse model in 1994, scientists quickly put their finger on a potential contributor to the disease: a power outage in the motor nerves. Researchers, led by Northwestern University School of Medicine neuroscientist Mark Gurney MD, now at Michigan’s Tetra Discovery Partners, reported that the mitochondria swelled up in the motor neurons of SOD mice before showing any signs of ALS suggesting that malfunctions in these intracellular power plants might, in part, lead to the disease.
“But whether [defective] mitochondria were driving the pathology of ALS, that was a question mark,” explains Johns Hopkins University School of Medicine neuroscientist Lee Martin PhD.
In 2004, University of California San Diego researchers led by neuroscientist Don Cleveland PhD found that these misshapen mitochondria appeared at the nerve terminals at about the same time as the muscles became unplugged.
Healthy
Mutant SOD1
Tracking Numbers. Researchers tracked the deliveries of mitochondria in healthy and mutant SOD1 cultured spinal cord motor neurons. Courtesy of Jordi Magrané, Weill Cornell Medical College. All Rights Reserved.
University of California San Diego neuroscientist Christine Vande Velde PhD, now at the University of Montreal, however suspected that more than the breakdown of these power plants could be contributing to ALS. She noticed that these swollen mitochondria accumulate at the neuromuscular junctions in these mice as the disease progressed suggesting that these power plants were unable to travel back towards the cell body to be refurbished or recycled. This so-called retrograde transport of mitochondria is critical to meet energy demands in power-guzzling regions of the motor nerves including the nerve terminals where electric signals are transmitted across the neuromuscular junction which ‘tell’ the muscles to move. If these signaling systems are on the fritz, this too could contribute to muscle weakness and paralysis.
To try and determine whether this impaired mitochondrial dynamics could also be contributing to ALS, Jordi Magrané PhD and Giovanni Manfredi PhD at Weill Cornell Medical College in New York introduced a system in the late 2000s in which they could fluorescently tag these intracellular power plants in cultured ALS SOD1 mouse spinal cord motor neurons in laboratory dishes and watch them move live in real time under a microscope.
Reporting just last month, the team found that mitochondrial trafficking is indeed affected in the SOD1 mutant motor neurons of the spinal cord. The number of mitochondria that undergo fusion – critical to keep these intracellular power plants in working order – dropped over 50%. And, the movement of mitochondria slowed to a crawl. (Check out the video.)
Incredibly, the Weill Cornell team found that newly generated power plants constructed in the cell body are already operating at reduced capacity and accumulate at the so-called distal end of the axon, near the nerve terminal. And, the number of mitochondria appear to be reduced at synapse-like structures. These defects appear to be specific to motor neurons.
“This could be happening in ALS,” says Magrané.
These findings come at the heels of a study from Vande Velde’s team last summer in which they discovered that mitochondria pile-up in the spinal cord in mutant SOD1 mice during the disease course.
Now, the Weill Cornell team is using the same tools to monitor these intracellular power plants in living ALS mice to determine whether mitochondrial dynamics is indeed defective and contributes to denervation. Looking ahead, the researchers hope to take a look at other mouse models of ALS to determine whether these multi-mitochondrial pile-ups generally contribute to the disease.
Tracking down the culprit
What causes the transport and recycling machinery to fail in ALS? Researchers remain unsure. One possibility is that the SOD1 enzyme which accumulates during the course of the disease could jam up the works. Reporting last summer, Vande Velde’s team found that these misfolded proteins stick to the surfaces of mitochondria in the spinal cord of ALS mice. This could make these intracellular power plants more difficult to pick up by motor proteins, the neurons’ delivery vehicles, and therefore more difficult to transport.
But new findings from the labs of Hugo Bellen PhD at the Baylor College of Medicine in Texas and Michael Miller PhD at the University of Alabama at Birmingham suggest that there could be an even bigger problem. The systems that keep these power plants upon delivery fixed in position and in tip-top condition could also be out to lunch.
Reporting last month, the Bellen-Miller team discovered that the putative hormone VAP-B produced by neurons fine-tunes the positions of mitochondria and regulates the energy production in muscles. A hormone that is lacking in all people with ALS tested including those with the sporadic form of the disease.
The Electric Slide
Scientists first stumbled upon a link between VAP-B and ALS in 2004 when geneticist Mayana Zatz PhD and colleagues at São Paulo University in Brazil reported that a mutation in the gene, also known as ALS8, triggered the disease. Reporting in 2008, the Bellen-Miller team discovered that VAP-B appears to be a hormone and that this mutation blocked its secretion. But why a drop in VAP-B levels resulted in ALS remained unclear.

Power punch. Muscles are packed with mitochondria (arrows) to generate the energy needed for contraction. Here, a section of skeletal muscle is shown. Image: NHBLI, NIH.
To try and get a better idea of why a lack of VAP-B could contribute to ALS, Bellen’s team at Baylor College of Medicine generated fruit flies unable to produce the hormone and watched them develop. The researchers found that very few of these flies survived but the few that did could barely move. Taking a closer look at their muscles, the Bellen team quickly identified the problem. Most of the mitochondria appeared to be broken down. Their muscles just simply did not have enough fuel.
But, these studies were extremely difficult to do. Too few flies survived for more detailed analysis. So, the researchers turned to Miller’s team at the University of Alabama who were studying the loss of VAP-B in the roundworm. The researchers also noticed that the mitochondria appeared to be malfunctioning. But they also noticed something else. The mitochondria were displaced from the muscle fibers. And, by watching the worms crawl under the microscope, the researchers found out why: these intracellular power plants were not fixed into place.
VAP-B, however, appears to do a lot more than make sure that mitochondria are next to muscle fibers ready to rock and roll. The hormone appears to regulate the maintenance (fusion) of these power plants and even the amount of fuel produced. Without VAP-B, the mitochondria in these worms’ muscles were operating at as low as 50% capacity.
”The muscles suffer due to a lack of energy,” explains Bellen. “They produce lactic acid. If you do that chronically, your muscles start to waste.”
Bellen suspects however, that the loss of this potentially critical hormone could be responsible for a lot more than muscle atrophy in people with ALS. Without mitochondria in the right places in the muscle, calcium that gets generated during movement can build up triggering twitching. And, the drop of a critical protein produced by muscles according to studies in fruit flies could lead to synaptic die-back.
Actin Up
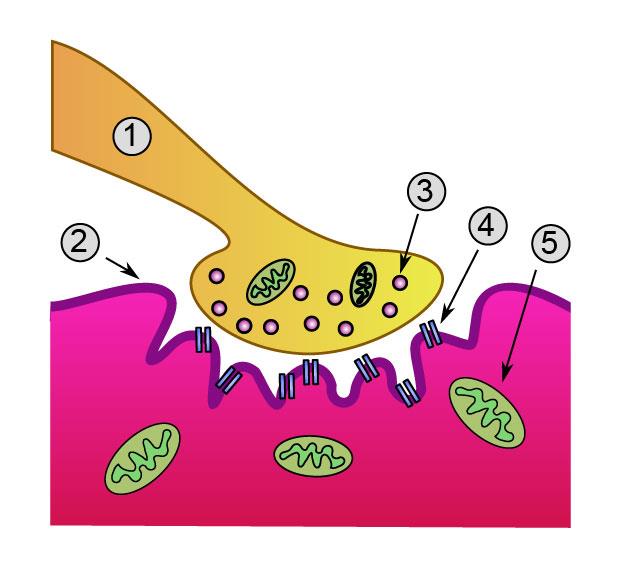
Junction box. Researchers discovered that VAP-B regulates signalling mechanisms which during development, help plug nerves into the right muscles. The team suspects that VAP-B helps enable the movement of these muscles (2) by stabilizing mitochondria (5) at the nerve terminal (1). Image: Wikimedia Commons.
Now, the researchers are looking to see whether a drop in VAP-B levels also results in reduced energy production in motor neurons and a loss of mitochondria from the nerve terminals.
VAP-B appears to be in the right place at the right time to control mitochondrial dynamics in the motor nerves. The hormone binds to receptors that are also present on the surface of adult nerve cells including motor neurons.
Furthermore, VAP-B appears to be able to do the job. The hormone controls the position of mitochondria in muscles according to the Bellen-Miller team's findings by regulating the length of the actin-based filaments that anchor them on muscle fibers. The same kinds of cytoskeletal fibers that are also present at nerve terminals.
“Microtubules form the freeway along which the mitochondria travel to the synapse. But once they reach the synapse,” explains Bellen,” they need to transfer to another transport system.”
And, that transport system could be controlled by VAP-B. VAP-B based signaling machanisms could stabilize these intracellular power plants at the nerve terminals – ensuring our muscles have the ability to move.
Looking ahead, the Bellen-Milller team hopes to figure out what triggers the release of VAP-B. By identifying these regulatory signals, researchers may be able to boost the production of the hormone in people with ALS and in so doing, slow down the disease.
But there may be no need to wait that long. VAP-B, in its secreted form, might be able to be administered directly to help keep the energy flowing in people with ALS.
“Maybe there is some therapeutic potential here,” says Miller. “But it is early days.”
References
Han, S.M, Tsuda, H., Yang, Y., Vibbert, J., Cottee, P., Lee, S.J., Winek, J., Haueter, C., Bellen, H.J., Miller, M.A. (2012) Secreted Vap8/als8 major sperm protein domains modulate mitochondrial localization and morphology via growth cone guidance receptors. Developmental Cell 22, 1-15. Abstract | Full Text (Subscription Required)
Magrane, J., Sahawneh, M.A., Przedborski, S., Estevez, A.G. and Manfredi, G. (2012) Mitochondrial dynamics and bioenergetics dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons. Journal of Neuroscience 32(1), 229-242. Abstract | Full Text (Subscription Required)
Cudkowicz, M., et al. (2011) The effects of dexpramipexole (KNS-760704) in individuals with amyotrophic lateral sclerosis. Nature Medicine 17(12), 1652-1656. Abstract | Full Text (Subscription Required)
Vande Velde, C, Garcia, M.L., Yin, X., Trapp, B.D. and Cleveland, D.W. (2004) The neuroprotective factor Wlds does not attenuate mutant SOD1-mediated motor neuron disease. NeuroMolecular Medicine 5(3), 193-203. Abstract | Full Text (Subscription Required)
Magrané, J., Hervias, I., Henning, M.S., Damiano, M., Kawamata, H., and Manfredi G. (2009) Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Human Molecular Genetics 18(23), 4552-4564. Abstract | Full Text
Vande Velde, C., McDonald, K.K., Boukhedimi, Y., McAlonis-Downes, M., Lobsiger, C.S., Bel Hadj, S., Zandona A., Julien, J.P., Shah, S.B. and Cleveland, D.W. (2011) Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS One 6(7), e22031. Abstract | Full Text
Nishimura, A.L., Mitne-Neto, M., Silva, H.C., Oliveira, J.R., Vainzof, M. and Zatz, M. (2004) A novel locus for late onset amyotrophic lateral sclerosis/motor neurone disease variant at 20q13. Journal of Medical Genetics 41(4), 315-320. Abstract | Full Text
Tsuda, H et al. (2008) The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell 133(6), 963-977. Abstract | Full Text
Goold, C.P. and Davis, G.W. (2007) The BMP ligand Gbb gates the expression of synaptic homeostasis independent of synaptic growth control. Neuron 56(1), 109-123. Abstract | Full Text
Ratnaparkhi, A., Lawless, G.M., Schweizer, F.E., Golshani, P. and Jackson, G.R. (2008) A Drosophila model of ALS: human ALS-associated mutation in VAP33A suggests a dominant negative mechanism. PLoS One 3(6), e2334. Abstract | Full Text